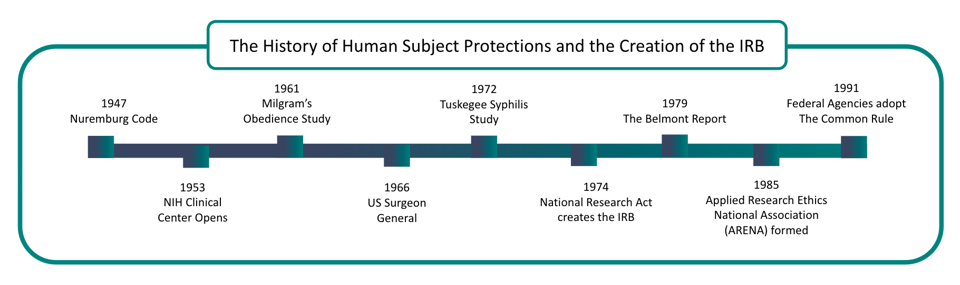
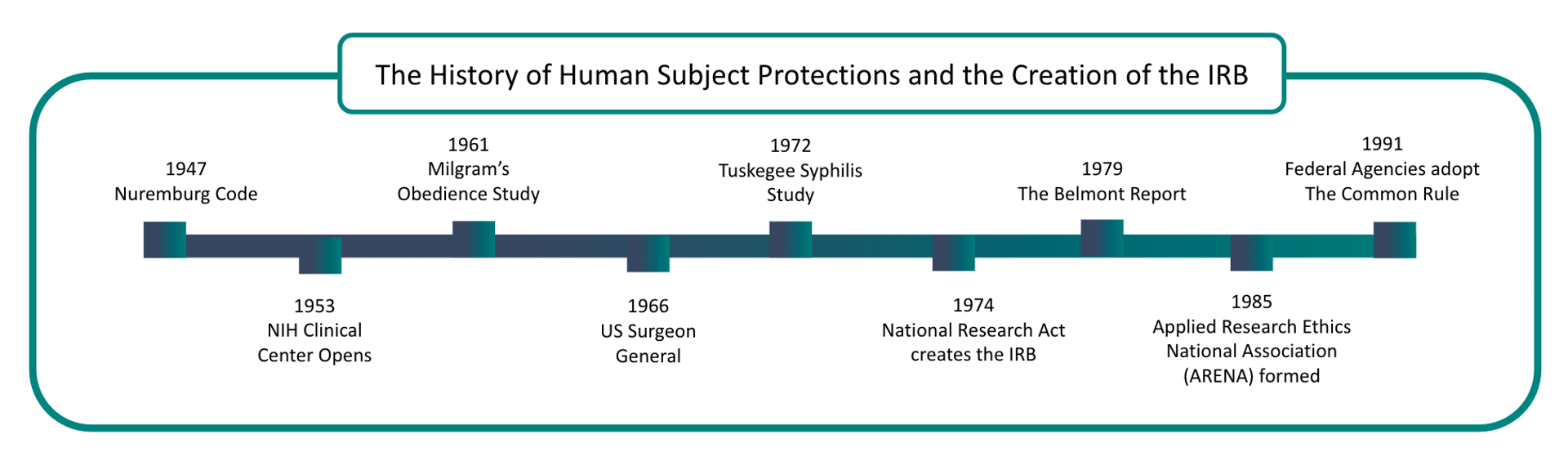
Medical Experimentation Abuse Comes to Light
The Nuremberg trials brought the abuse by World War II Nazi doctors in medical experimentation to light. Authorities recognized the need for guidelines for treating human subjects in clinical research.
As a result, in 1945, the Nuremberg Code became the first legal effort to address ethical research concerns.
Requiring Research Approvals
In 1953, the National Institute of Health (NIH) required all clinical research to be approved.
The United States Public Health Service expanded this to all “extramural” research in 1966 with new regulations applying to all agency-supported efforts.
Establishing Institutional Review Boards After Continued Unethical Treatment
In 1972, the Tuskegee Syphilis Study came to light. This study allowed 300 black rural men to go untreated for diagnosed syphilis even after antibiotics became available to treat this infection.
This study lead to the establishment of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research.
In the following years, revised regulations established the first Institutional Review Boards at hundreds of federally funded research institutions.
The Belmont Report
The Belmont Report, released in 1979, inspired revised regulations in the Federal Register.
The National Commission created the Belmont Report for the Protection of Human Subjects of Biomedical and Behavioral Research. It was written based on three core principles: respect for persons, beneficence, and justice.
These principles remain the basis for the United States Department of Health and Human Services (HHS) human subject protection regulation. Today, the Belmont Report is an essential reference for IRB review of human subjects research.
IRBs and the FDA
The U.S. Food and Drug Administration (FDA) regulates research involving drugs, biologics, and medical devices. It has established specific requirements for the review and approval of such research.
These requirements are outlined in the Code of Federal Regulations and include provisions for the ethical conduct of research involving human subjects.
To conduct a research study involving drugs, biologics, or medical devices, an IRB registered with the FDA must approve it. The IRB must review and approve the research protocol and ensure it meets ethical standards and complies with relevant laws and regulations.
Each study participant must sign an IRB-approved consent form before completing any study procedures. The IRB must also ensure that informed consent is clearly written and that the consent explains all procedures to be completed throughout the study.
The FDA also guides IRBs and researchers on ethical and regulatory issues related to research involving drugs, biologics, and medical devices. This guidance helps ensure that such research respects the rights and welfare of human subjects.
Any research conducted or supported by HHS follows leadership from the Office for Human Research Protections (OHRP) to protect human subjects’ rights, welfare, and well-being. OHRP also ensures that the research follows law 45 CFR 46 (Protection of Human Subjects).
IRB Membership
Members of an Institutional Review Board (IRB) are typically drawn from a variety of disciplines and backgrounds. IRB members may include scientists, medical professionals, lawyers, ethicists, and laypeople without specific scientific or medical training.
Particular IRB composition depends on the research and the organization’s needs. However, there are minimum board member requirements that the CFR sets forth.
In general, IRB members should:
- Understand ethical principles and federal regulations related to research involving human subjects
- Apply these principles and regulations to the review of research protocols
- Effectively communicate their concerns and recommendations to researchers and other members of the IRB
In addition to the primary members of the IRB, there may also be alternate members who can participate in the review process if a primary member cannot attend a meeting or is otherwise unavailable. There may also invite ex-officio members to participate in the review process as needed, based on their specific expertise or experience.
Institutional Review Board Review Processes
The typical review process for an Institutional Review Board (IRB) involves the following steps:
1. Submission of a Research Protocol
Researchers who wish to conduct research involving human subjects must submit a detailed research protocol to the IRB for review. The protocol should include information about the:
- Research question
- Study design
- Planned patient sample
- Methods for recruiting and consenting subjects
- Procedures
- Risks or benefits associated with participation
2. Review of the Protocol
The IRB review the research protocol to ensure that it:
- Meets ethical standards
- Complies with relevant laws and regulations
- Offers the currently accepted standard of care for the studied indication
The IRB may ask the researcher to modify the protocol or provide additional information to address any concerns raised during the review process.
3. Review of the Informed Consent
The informed consent process is how the study coordinator or investigator communicates the details about the research study to the potential subject. Informed consent is a process to help ensure the potential research subject understands the study details.
The fundamental purpose of the IRB review of informed consent is to ensure that subjects’ rights and welfare are protected and that they are making a fully informed decision.
4. Approval of the Protocol
If the IRB determines that the protocol meets ethical standards and complies with relevant laws and regulations, it will approve the protocol and allow the research to proceed. If the IRB has concerns about the protocol, it may request additional information, request modifications, or decide to deny approval of the study.
5. Ongoing Review
The IRB will also conduct an ongoing review of the research project to ensure it meets ethical standards and complies with relevant laws and regulations. This involves reviewing progress reports and may include conducting site visits and reviewing any changes to the protocol that the researcher proposes.
A review must occur at least annually when a study is active. However, an IRB may stipulate more frequent reviews if the Board decides it’s necessary due to the nature of the study.
The Importance of an Institutional Review Board in Clinical Research
One of the main functions of the IRB is to balance the interests of moving science forward while protecting the subject. IRBs serve as an essential safeguard for patient safety by confirming that research involving human subjects is conducted ethically and in a manner that respects the rights and welfare of the subjects. If a clinical trial knowingly jeopardizes a patient’s well-being or is conducted in an unethical manner, the risks would outweigh the potential benefits of conducting the study.
The profound good that continues to be done by clinical research teams is fostered and continuously supported by strong and efficient Institutional Review Boards. To learn more about IRBs and their value to all stakeholders in clinical research, contact the NJ, NY, and PA clinical trial experts at Vitalief.